Flowchart of the new method in cellular structural biology
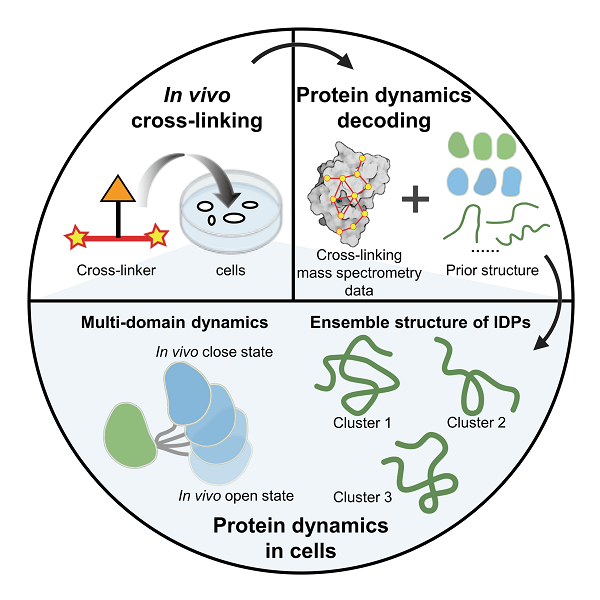
The research team first proposed a structural reconstruction strategy for multi-domain proteins. There are rich dynamic changes between different structural domains of multi-domain proteins, and these dynamic changes are difficult to capture by conventional structural biology methods and structural prediction algorithms. The research team proposed to treat individual structural domains as a whole and optimize the overall three-dimensional structure using the distance information corresponding to the XL-MS data between the structural domains. Using this overall reconstruction strategy, the dynamic structures of calmodulin, hnRNP A1, and hnRNP D0 proteins in cells were successfully characterized. Naturally disordered proteins have important biological functions, but their high dynamics make it difficult for traditional structural biology methods to resolve their structures, and AlphaFold2 cannot accurately predict them. In response to this problem, the research team proposed two complementary structural characterization strategies: one is to directly convert XL-MS information into distance constraints for the calculation of naturally disordered protein structures; the other is to first use all-atom molecular dynamics simulations for unbiased sampling, and use XL-MS data-based evaluation and screening of sampled structures. Using these two complementary research strategies, the team successfully decoded the ensemble conformations of two highly mobile histones HMG-I/Y and HMG-17 in cells. These two complementary structural characterization strategies not only achieve comprehensive sampling of molecular conformations of naturally disordered proteins, but also ensure the accuracy and reliability of results through structural clustering and re-evaluation methods, avoiding possible overfitting states, thereby comprehensively and objectively reconstructing the ensemble conformation of naturally disordered proteins in cells. This study provides key tools and research ideas for a deeper understanding of protein function in cells.
The first author of this paper is Zhang Beirong, a doctoral student at the Dalian Institute of Chemical Physics, and Gong Zhou, an associate researcher at the Institute of Precision Measurement. The corresponding authors are Zhang Lihua and Zhao Qun, researchers at the Dalian Institute of Chemical Physics, and Gong Zhou at the Institute of Precision Measurement.
This work was supported by major projects of the National Natural Science Foundation, the National Key R&D Program, and the Chinese Academy of Sciences Youth Promotion Association.
Full text link:https://doi.org/10.1002/anie.202301345